Uzun QT Sendromu
Peter J. Schwartz, M.D., and Lia Crotti, M.D., Ph.D.
N Engl J Med 2025;393:2023-34.
Sendrom, hastanın istirahat hâlinde çekilen elektrokardiyogramında (EKG) QT aralığının uzaması ve çoğunlukla fiziksel ya da duygusal stres koşullarında ortaya çıkan, yaşamı tehdit eden aritmilere yatkınlık ile karakterizedir.5,7
Sendromun zamanında tanısının klinik önemi, ani kardiyak ölümün çoğu zaman ilk belirti olabilmesinden kaynaklanır; bu durum, tanısal veya terapötik hataların telafi edilmesini imkânsız hâle getirir. Bundan 50 yıl önce belirtildiği gibi,5 günümüzde mevcut tedavilerin yüksek etkinliği göz önüne alındığında, tanı konmamış — dolayısıyla tedavi edilmemiş — uzun QT sendromlu hastalara tanı konulamamış olması artık affedilemez bir durumdur; ne yazık ki, atlanan tanılar hâlâ çok sık görülmektedir.
Uzun QT Sendromunun Genetik Temeli
Uzun QT sendromu ile ilişkili üç ana gen (vakaların yaklaşık %90’ında görülür) — KCNQ1, KCNH2 ve SCN5A — 1995 ve 1996 yıllarında tanımlanmıştır.8-10
KCNQ1 ve KCNH2 genlerindeki varyantlar sırasıyla hastaların yaklaşık %50’sinde tip 1 uzun QT sendromunun ve %40’ında tip 2 uzun QT sendromunun nedenidir; bu genler, dışa doğru akımları olan IKs ve IKr’yi ileten potasyum kanallarını kodlar. Bu kanallar kardiyak repolarizasyon için kritik öneme sahiptir ve patojenik varyantların neden olduğu IKs ve IKr akımlarındaki azalma QT aralığını uzatarak uzun QT sendromuna yol açar.11 Adrenerjik aktivasyon sırasında — örneğin fiziksel aktivite esnasında — IKs akımı baskın repolarizasyon akımı hâline gelir ve bu değişikliğin önemli klinik sonuçları vardır: kalp hızı arttığında QT aralığı uygun şekilde kısalmazsa ventriküler fibrilasyon gelişebilir.
Üçüncü ana gen olan SCN5A, başlıca depolarizan içe doğru sodyum akımı olan INa’yı ileten voltaj kapılı sodyum kanalını kodlar. SCN5A’nın "gain of function" (işlev artışı) oluşturan patojenik varyantları repolarizasyonu uzatarak vakaların yaklaşık %10’unda tip 3 uzun QT sendromuna neden olur.
KCNQ112 ve KCNE113 (IKs potasyum kanalının alt birimlerini kodlayan) genlerindeki homozigot veya bileşik heterozigot patojenik varyantlar, konjenital sağırlık ile ilişkili resesif Jervell–Lange-Nielsen sendromuna yol açar.2,14
Uzun QT sendromu ile ilişkilendirilen ek genler tanımlanmış olsa da bunlardan yalnızca birkaçının önemli rolü vardır.15 Kalsiyum kanalı geni CACNA1C’deki varyantlar, uzun QT sendromunun bir formu olan ve iskelet ile nörogelişimsel anormalliklerin eşlik ettiği Timothy sendromuna neden olur.16
CALM1, CALM2 ve CALM3 genlerindeki varyantlar — başlıca kardiyak iyon kanallarını modüle eden kalmodulini kodlayan genlerdir — yaşamı tehdit eden aritmiler ve diğer kardiyak veya nonkardiyak patolojik özelliklerle ilişkili kalmodulinopatilere yol açar.18,19
Kalmodulin genleri benzer değillerdir; farklı kromozomlarda bulunurlar ancak aynı proteini kodlarlar. Bu üç gendeki patojenik varyantlar, kalsiyum kanal inaktivasyonunu bozarak17 QT aralığını uzatır ve uzun QT sendromuna neden olur.
Uzun QT sendromunda genetik analizlerde klinik anlamı belirsiz varyantlar (VUS) sıkça saptanır ve bu durum hastayı takip eden hekim için kafa karıştırıcı olabilir. Bu tür varyantlar, artan bilgi birikimiyle — genellikle klinik fenotipin belirginliği,20,21 aile bireylerinde birlikte görülme kanıtı (kosegregasyon) veya fonksiyonel değerlendirme22 sayesinde — periyodik olarak yeniden sınıflandırılır. Böylece “benin”, “muhtemelen benin”, “patojenik” veya “muhtemelen patojenik” kategorilerine ayrılırlar;23 bu sınıflandırmalar çoğu zaman klinik yönetimi de etkiler.
Uzun QT sendromu esas olarak monogenik bir hastalık olmasına rağmen, yaygın genetik varyantların toplam etkisi (poligenik risk skorları ile temsil edilir) özellikle genotip negatif hastalarda sendroma duyarlılığı modüle edebilir.24,25
Değiştirici (Modifiye Edici) Genler
Güney Afrika’da geniş bir popülasyonunda,26 aynı varyantı (KCNQ1–A341V) taşıyan yüzlerce birey arasında kalp hızı için düzeltilmiş QT aralığında (QTc) geniş bir dağılımın bulunması, uzun QT sendromunda değiştirici genlerin tanımlanması ve incelenmesi için benzersiz bir fırsat sunmuştur.27
Değiştirici genler terimi, genellikle yaygın olan ve hastalık oluşturan varyantların sonuçlarını her iki yönde — artırarak veya azaltarak — değiştirme kapasitesine sahip genetik faktörleri ifade eder.27
Son 20 yıl içinde birkaç değiştirici gen tanımlanmış olup,27-29 bunun iki ana klinik sonucu vardır:
- Risk sınıflandırmasının daha hassas hâle getirilmesi:
Bu genler, hastanın risk düzeyine göre daha agresif veya daha az agresif tedavi stratejilerinin belirlenmesini sağlar. - Yeni tedavi yaklaşımlarının geliştirilmesi:
Değiştirici genlerin uzun QT sendromunda ortaya koyduğu hücresel etki mekanizmaları,27,29,30 belirli moleküler yolakları hedefleyen yeni tedavilerin tasarlanmasının önünü açmaktadır.
Klinik Bulgular ve Tanı
Uzun QT sendromunun temel özellikleri EKG ve aritmik olaylara ilişkindir. QT aralığı genellikle belirgin şekilde uzundur ve sıklıkla ventriküler repolarizasyonla ilişkili alışılmadık morfolojik değişiklikler (örneğin, bifazik ve çentikli T dalgaları) ile birliktedir; bu bulgular, ölçüm yapılmadan önce bile tanısal şüphe uyandırmalıdır. Nitekim uzun QT sendromu söz konusu olduğunda, patern tanıma son derece önemlidir (Şekil 1 ve Tablo 1).
Şekil 1
Bazı elektrokardiyografik (EKG) paternler, QT aralığının gerçek uzunluğundan bağımsız olarak uzun QT sendromunu düşündürür.
A

A-Normal bir EKG ve kalp hızına göre düzeltilmiş QT aralığını (QTc) 417 msn olarak göstermektedir.
B
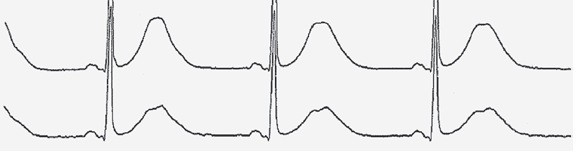
B- Geniş tabanlı T dalgalarını, T dalgası çentiklerini ve 615 msn’lik bir QTc’yi göstermektedir.
C

C -iki fazlı T dalgalarını ve 577 msn’lik bir QTc’yi göstermektedir.
D
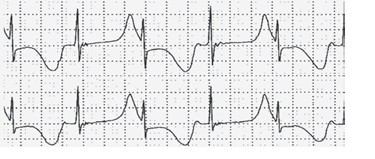
D- uzun QT sendromunun tipik bir EKG özelliği ve yüksek elektriksel instabilite göstergesi olan T-dalgası alternansını ve 776 msn’lik bir QTc’yi göstermektedir.
E
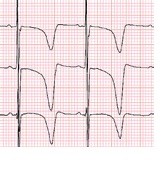
E- derin negatif T dalgalarını ve 673 msn’lik bir QTc’yi göstermektedir.
F
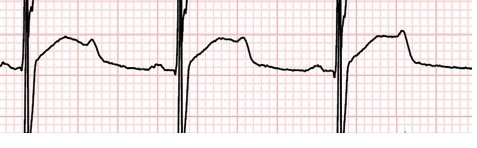
F-uzun QT sendromu tip 2 için tipik olan çentikli T dalgalarını ve 483 msn’lik bir QTc’yi göstermektedir.
G
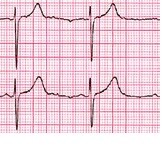
G- İstirahat EKG.G ve Panel H aynı hastaya aittir.
H
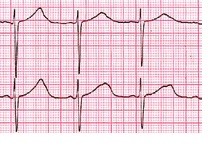
H- egzersiz stres testinin sonunda toparlanma fazında 640 msn’lik QTc uzamasını (H), başlangıç (G) QTc değeri olan 472 msn ile karşılaştırmalı olarak göstermektedir.
|
Tablo 1. Uzun QT sendromu için tanı kriterleri LQTS (uzun QT sendromu) |
|
Puanlama Kriterleri |
|
Toplam puanlar LQTS olasılığını şu şekilde göstermektedir: 0–1 puan, düşük olasılık 1,5–3 puan, orta düzey olasılık 3,5 puan ve üzeri, yüksek olasılık |
|
Elektrokardiyografi (Elektrokardiyografik veriler, QT aralığını uzatan ilaç kullanmayan ve böyle bir durumu olmayan hastalardan elde edilmelidir) |
|
QTc (Kalp hızına göre düzeltilmiş QT aralığı (QTc), Bazett’in formülüne göre hesaplanır27)
Torsades de pointes 2 puan (Torsades de pointes ile senkop birbirini dışlar; Anlam olarak: Aynı anda birlikte görülmezler / biri varsa diğeri tanı kriteri olarak kabul edilmez.) T-dalga alternansı 1 puan Üç derivasyonda çentikli T dalgası 1 puan Yaşa göre kalp hızının düşük olması 0,5 puan (yaşa göre düşük kalp hızı, istirahat kalp hızının yaş için %2 persentilin altında olması olarak tanımlanır) |
|
Klinik |
|
Senkop (Torsades de pointes ile senkop birbirini dışlar; Anlam olarak: Aynı anda birlikte görülmezler / biri varsa diğeri tanı kriteri olarak kabul edilmez.)
Konjenital sağırlık 0.5 puan Aile hikayesi (aynı aile bireyi iki kere hesaba katılmamalıdır) ≥ Bir den fazla kanıtlanmış LQTS lu aile bireyi olması 1 puan Otuz yaşından küçük bir birinci derece aile üyesinde açıklanamayan ani kardiyak ölüm 0,5 puan |
Bazett formülüne göre kalp hızına düzeltilmiş QT (QTc) değerinin normal üst sınırları erkekler için 440 ms ve kadınlar için 460 ms’dir (Bazett formülüne göre kalp hızı düzeltilmesi ile31). Sınırlılıklarına rağmen, Bazett formülüne göre yapılan düzeltme, normal ve anormal değerleri — bebeklerde bile — yararlı biçimde ayırt eder.32 QT aralığı, Q dalgasından T dalgasının taban çizgisine döndüğü noktaya kadar ölçülmelidir; zaman kazandırdığı için yaygın olarak kullanılan tanjant yöntemi, ventriküler repolarizasyonun gerçek süresini çoğu zaman olduğundan kısa tahmin ederken, en uzun QT aralığı, aritmik risk değerlendirilirken dikkate alınması gereken en önemli değerdir.33 QTc’nin 500 ms’nin üzerinde olması, orta veya yüksek aritmik risk altındaki hastaları ayırt etmeye yardımcı olur.34 T dalgasındaki çentikler (Şekil 1), sıklıkla mekanik değişikliklerle birlikte görülür,35-37 erken ardıl depolarizasyonlara bağlı olarak aritmi riskinin bir göstergesidir38 ve özellikle uzun QT sendromu tip 2 hastalarında daha sık izlenir. T-dalgası alternansı (Şekil 1), kedilerde sol stellat ganglionun uyarılmasıyla QT uzamasıyla birlikte deneysel olarak yeniden oluşturulmuş olup,39 ventriküler fibrilasyondan önce görülen önemli bir belirti ve ciddi kardiyak elektriksel instabilite göstergesidir. T dalgası morfolojik özellikleri belirli genotiplerin öngörülmesine yardımcı olabilir, ancak gerçek genetik taramanın yerine geçemez. Aritmik olaylar, sıklıkla ventriküler fibrilasyona ilerleyen torsades de pointes tipi ventriküler taşikardi sonucu ortaya çıkar ve bu durum kalp durmasına ve ani ölüme yol açabilir. Semptomlar ve klinik sonuç, torsades de pointes’in süresine bağlıdır. QT uzaması olan bireylerde kısa süreli senkop veya vertigo gelişmesi, torsades de pointes olasılığı açısından hekimi uyarmalıdır; zira bu durum çoğu zaman hayatı tehdit eden aritmik epizotların habercisidir. Uzun QT sendromunda aritmik olaylara yol açan gen-özgül tetikleyiciler tanımlanmıştır40. Uzun QT sendromu tip 1 olan kişilerde, özellikle yüzme sırasında olduğu gibi duygusal veya fiziksel stresler sırasında sempatik aktivitenin arttığı durumlarda risk artar40. Uzun QT sendromu tip 2 olan kişilerde ise ani gürültüye maruziyet, özellikle dinlenme ya da uyku sırasında aniden uyanmaları halinde riski artırır40; ayrıca bu hastalar düşük plazma potasyum düzeylerine ve QT aralığını uzatan ilaçlara son derece duyarlıdır ve kadın hastalarda postpartum dönemde — muhtemelen uykunun bölünmesine bağlı olarak aritmojenik REM uykusu reboundu nedeniyle — risk yüksektir. Uzun QT sendromu tip 3 olan kişilerde ise risk esas olarak dinlenme veya uyku sırasında ortaya çıkar (Tablo 2).
Tablo 2- Genotip özgül tedavi *
|
Tedavi yönetimi |
LQT1 |
LQT2 |
LQT3 |
|
Yanıt † |
|||
|
Beta Blokör |
+++ |
++ |
++ |
|
Sempatik Denervasyon |
+++ |
++ |
++ |
|
Mexiletin |
Bilinmiyor |
++ |
+++ |
|
Tetikçiler veya eşlik eden olaylar |
Adrenerjik- yoğun egzersiz, Yüzme, yoğun duygusal olaylar |
Ani ve yüksek seslerle ortaya çıkan irkilme (ör. çalar saat, telefon zil sesi), düşük serum potasyum düzeyi ve doğum sonrası dönem |
Uyku veya dinlenme |
|
Öneriler |
Ağır egzersizden kaçınılmalı (yüzmeye, yüzme bilen bir yetişkin gözetiminde izin verilebilir), sözlü veya fiziksel çatışmalardan uzak durulmalı ve riskin yeniden değerlendirilmesi için yıllık takip ziyareti yapılmalıdır |
Serum potasyum düzeyi ≥4 mmol/L olacak şekilde korunmalıdır. Yatak odasında çalar saat ve telefon kullanılmamalıdır. Beta-blokerler sabah ve akşam alınmalıdır. Doğum sonrası dönemde uyku güvenliği için eşle aynı odada kalınması önerilir‡. Riskin yeniden değerlendirilmesi için yıllık kontrol ziyareti yapılmalıdır |
Ev tipi otomatik eksternal defibrilatör‡ ve eşle/başkasıyla aynı odada kalmanın§ potansiyel yararı vardır; riskin yeniden değerlendirilmesi için yıllık takip ziyaretleri önerilir. |
Tablo-2 *LQTS tip 1 (LQT1), aritmilerin fiziksel veya duygusal stres sırasında gelişme eğilimi ile karakterizedir; tip 2 (LQT2), aritmilerin özellikle kişi istirahat halindeyken yüksek seslere maruz kalma sonrasında ve uyku düzeninin bozulmasını takiben gelişme eğilimi ile karakterizedir; tip 3 (LQT3) ise aritmilerin kişinin istirahat halinde veya uykuda olduğu sırada gelişme eğilimi ile karakterizedir. Bunun istisnaları mevcuttur.
† Yanıtın büyüklüğü, + sembolleriyle gösterilmiştir ve + (en düşük yanıt büyüklüğü) ile +++ (en yüksek yanıt büyüklüğü) arasında değişmektedir.
‡ Şiddetli olgularda evde otomatik eksternal defibrilatöre erişim önemli olabilir, çünkü olayların çoğu kişi istirahat halindeyken veya uykudayken meydana gelmektedir.
§ Uyku sırasında yatay pozisyon ve ventriküler taşikardi ile ventriküler fibrilasyon sırasında oksijen perfüzyonunun giderek azalması göz önüne alındığında, çoğu hastanın çevredekilerin hızlı resüsitasyon yapabilmesine imkân veren boğuk, derin hırıltı gibi agonik sesler çıkarabilecek zamanı olmaktadır.
Genotipten bağımsız olarak, yaşamın ilk yılında kardiyak olay geçiren bebekler çok yüksek ölüm riski altındadır ve geleneksel tedavilerle nadiren korunurlar.41 Uzun QT sendromu, bebeklik dönemindeki ani ölüm sebeplerinden biridir42. İlk yıl içinde ani ölümle kaybedilen bebeklerin %10’una kadarında43 veya uterusta kaybedilen fetuslarda44 uzun QT sendromuna neden olan varyantlar saptanmıştır ve bu bulgulara göre yeni doğanlarda uzamış QTc, ani ölüm riskini artırır45. Genetik test yapılmadığında, yaşamın ilk aylarında kaybedilen bebeklerin ani ölümü ani bebek ölümü sendromu olarak isimlendirilir. Bu aşırı basitleştirici yaklaşım yerine, yaşamın ilk ayında EKG taraması yapılması daha akılcı bir yaklaşımdır46 böylece,ölüm riski taşıyan uzun QT sendromlu bebekler tespit edilebilir46. Bu değerlendirmeler ayrıca, birden fazla kardeşte görülen bebeklik çağı ani ölümlerinin ihmal olarak varsayılmadan önce temkinli olunması gerektiğini de hatırlatır47.
Tipik olgularda örneğin belirgin QTc uzaması ile birlikte senkop ile gelen hastada tanı doğrudan konulabilir. Sınırda olgularda (örn. hafif QTc uzaması ve semptomsuzluk) genetik tarama yardımcı olabilir, ayrıca 12 derivasyonlu, 24 saatlik Holter kaydı, özellikle gece saatlerinde tipik değişiklikleri ortaya çıkarabilir. Efor testi sonrası iyileşme fazında QTc uzaması veya egzersizin zirvesinde T ve P dalgalarının tam füzyonunun ortaya çıkması48 tanıya katkı sağlayabilir. Her hekimden uzun QT sendromunu kesin olarak tanılaması beklenmez; ancak QT aralığı uzaması olan senkop olsun ya da olmasın — çocuk veya ergen ile karşılaşıldığında ve ikincil nedenler dışlandıktan sonra, sendromdan şüphelenilmeli ve hasta konu hakkında özel deneyimi olan bir merkeze yönlendirilmelidir. Uzun QT sendromu tanısında özel deneyimi olmayan hekimler için, yıllar içinde geliştirilen ve sendrom olasılığının ön değerlendirmesinde yararlı olan bir tanısal skor sistemi kullanışlıdır (Tablo 1)49 Uzun QT sendromu tanısını desteklemek amacıyla bazı testler önerilmiştir50. Bunlardan gerçekten yararlı olan tek test, egzersiz testidir; çünkü toparlanmanın (recovery) dördüncü dakikasında belirgin QT uzaması, uzun QT sendromu için yüksek özgüllüğe sahiptir49,51. Ayağa kalkma testi (stand-up test) sınırlı değere sahiptir52,50. Epinefrin provokasyon testi, genetik taramanın nadir bulunduğu dönemlerde önerilmiş olsa da53, tehlikeli aritmojenik potansiyele sahiptir ve normal EKG’si olan kişilerde bile ventriküler repolarizasyonu belirgin biçimde değiştirebilir; bu durum yanlış şekilde uzun QT sendromu izlenimi doğurabilir. Avrupa Kardiyoloji Derneği kılavuzuna göre54 epinefrin testi tanı amacıyla kullanılmamalıdır.
Tedavi
Tedavinin dört temel ayağı beta-blokerler, mexiletin, sol kardiyak sempatik denervasyon ve implante edilebilir kardiyoverter–defibrilatör (ICD)’dir. Bu tedaviler, uzun QT sendromunun altta yatan patofizyolojisine göre belirlenir. Buna ek olarak, QT’yi uzatan ilaçlardan kaçınma (bu ilaçların listesi şu adreste mevcuttur: https://www.crediblemeds.org/ ) ve potasyum takviyelerinin kullanılması (yeterli plazma potasyum düzeylerinin korunması için) gibi yaşam tarzı düzenlemeleri de aritmi riskinin azaltılmasına önemli ölçüde katkı sağlayabilir54.
Beta-Blokerler
1970’lerin ortalarından bu yana beta-blokerler, uzun QT sendromu olan hastalarda tedavinin temelini oluşturmuştur5,55 ve etkinlikleri, genotipten bağımsız olarak defalarca doğrulanmıştır7,56,57. Sendromda etkinliği kanıtlanmış iki beta-bloker propranolol (günlük 2,0–3,5 mg/kg) ve nadolol (günlük 1,0–1,5 mg/kg)’dür7. Metoprolol kullanılmamalıdır58. Beta-bloker tedavisindeki ciddi ve yaşamı tehdit eden başarısızlıkların çoğu, tedaviye uyumsuzluk ve QT’yi uzatan ilaç kullanımı ile ilişkilidir59. Beta-blokerler, genotip-pozitif fakat fenotip-negatif kişiler için de reçete edilmelidir,25 bazı gen-özgül istisnalar dışında (örn. tedavisiz olarak 25 yaşına kadar asemptomatik kalan uzun QT tip 1 erkek hastalar)40. Tanının belirsiz olduğu, sınırda QT uzaması olan genotip-negatif hastalarda karar vermek zordur; çünkü beta-bloker tedavisi başlandıktan sonra bunu kesmek çoğu zaman büyük ölçüde medikolegal nedenlerle güçleşir.
Sol Kardiyak Sempatik Denervasyon
Günümüzde çoğunlukla torakoskopik yöntemle uygulanan sol kardiyak sempatik denervasyon,60,61 Horner sendromunu önlemek için stellat ganglionun sadece alt yarısının61,62 ve T1–T4 torakal ganglionlarının çıkarılmasını içerir. Bu yöntemin etkinliği deneysel61 ve klinik kanıtlarla61-63 desteklenmektedir ve büyük ölçüde belirgin antifibrilatuvar etki,64 ventrikül düzeyinde norepinefrin salınımında belirgin azalma rağmen denervasyon sonrası aşırı duyarlılık gelişmemesi61 ve kalp hızında azalma olmaması61ile sağlanır. Geniş hasta serilerinde sol kardiyak sempatik denervasyon, son derece yüksek başarı oranı göstermiştir63,65,66 ve elektrik fırtınaları (çoklu VT/VF epizodları ve tekrarlayan ICD müdahaleleri) nedeniyle uygulandığında, yıllık ICD şok insidansını %90 oranında azaltmıştır63,65,67 ve böylece iyi bir yaşam kalitesinin korunmasını sağlamıştır68. Çoğu hastada klinik olarak anlamlı QTc kısalması görülür ve bu etki uzun dönem koruma sağlar63. Sonuç olarak, tam doz beta-bloker tedavisine rağmen senkop atakları devam eden hastalarda sol kardiyak sempatik denervasyon düşünülmeli ve tereddüt edilmeden uygulanmalıdır. Dünya genelinde bu işlemi gerçekleştiren merkezlerin sayısının giderek artması66,69,70 nedeniyle, bu hastalara sol kardiyak sempatik denervasyon ile ICD arasındaki avantaj ve dezavantajlar ayrıntılı biçimde anlatılmadan doğrudan ICD implantasyonu yapılması artık haklı gösterilemez71,72.
Mexiletin
1995 yılında, uzun QT sendromuna neden olan SCN5A varyantlarının sodyum akımını artırdığı keşfedildikten kısa bir süre sonra9,11, sodyum kanal blokeri olan mexiletin, uzun QT sendromu tip 3 için ilk gen-özgül tedavi olarak önerildi73 ve günümüzde bu hastalarda yaygın olarak kullanılmaktadır; temel amaç QTc’yi kısaltmak ve böylece aritmi riskini azaltmaktır74. Uzun QT sendromu tip 3 varyantlarının çoğu (fakat hepsi değil) mexiletine yanıt verir75. Yakın tarihli veriler, uzun QT sendromu tip 2 hastalarının neredeyse %70’inde mexiletin ile QTc’nin kısaldığını göstermektedir,76 bu da ilacın klinik kullanım alanını önemli ölçüde genişletmektedir. Etkisini değerlendirmek için akut oral ilaç testi kullanılır; bu testte mexiletin 6–8 mg/kg dozunda oral yoldan uygulanır ve 2 saat içinde QTc’de anlamlı bir kısalma (>40 ms) olup olmadığı değerlendirilir. Bu şekilde yalnızca mexiletine pozitif yanıt veren hastalarda uzun süreli tedaviye başlanır76.
ICD
ICD kullanımında dünya genelinde büyük farklılıklar bulunmaktadır;77 Amerika Birleşik Devletleri’ndeki bazı merkezler uzun QT sendromu olan hastalarının yaklaşık %50’sine ICD implante ederken, sendromlu hastaların büyük serilerini takip eden iki merkez (Mayo Clinic ve Istituto Auxologico Italiano, Genetik Kökenli Kardiyak Aritmiler Merkezi) ICD’yi yaklaşık %5 oranında implante etmektedir78.İntravenöz ICD, subkutan ICD’ye tercih edilir; çünkü pacing yapılmasına olanak tanır ve bu özellik, beta-bloker dozunun artırılmasının gerektiği çok düşük kalp hızı olan hastalarda veya aritmi fırtınaları sırasında durumlarda hayati önem taşır.Belgelenmiş kardiyak arrest sonrası, beta-bloker tedavisi olsun ya da olmasın ICD implantasyonu mantıklıdır. ICD yerleştirilen 233 uzun QT sendromu hastasını içeren bir çalışmada67 hastaların çoğunun kardiyak arrest geçirmediği ve ayrıca birçoğunda beta-bloker tedavisinin başarılı olduğunu göstermiştir. Tip 1 ve tip 2 gruplarında asemptomatik hastalar neredeyse yokken, tip 3 grubunda bu oran %45 olup, SCN5A’daki patojenik bir varyantın asemptomatik kişilerde bile ICD implantasyonu için yeterli görüldüğünü göstermektedir. Ortalama 5 yıllık takipte hastaların %25’inde advers olay görülmüştür. Uzun QT sendromu hastalarında ICD kullanımı aşırı düzeydedir ve bu hastaların çoğunun ICD’ye ihtiyaç duymayacaktır62. Yaklaşık 1000 hastaya ait veriler, yıllık tedavi optimizasyonu ile üçlü tedavi (beta-bloker, mexiletin ve sol kardiyak sempatik denervasyon) uygulandığında, ICD kullanımının minimum düzeyde tutulmasına rağmen hastaların neredeyse tümünün hayatta kalabildiğini göstermektedir56.
ICD implantasyonu şu durumlarda önerilmelidir:
- Uygun tedaviye uyumlu iken kardiyak arrestten sağ kurtulan hastalarda
- Tam doz beta-bloker tedavisine rağmen senkop olan ve sol kardiyak sempatik denervasyon ile mexiletine tedavisineerişimi olmayan hastalarda
- Tam doz beta-bloker ve sol kardiyak sempatik denervasyona rağmen senkopu devam eden tüm hastalarda.
Yeni Farmakolojik Tedaviler?
Deneysel kanıtlara dayanarak indüklenmiş pluripotent kök hücre kardiyomiyositleri ile yapılan çalışmalar79-81 uzun QT sendromu için uygulanmaktadır. Cesaret verici ön gözlemler sonucunda,82 kistik fibroz tedavisinde kullanılan lumakaftor–ivakaftor kombinasyonu, taşınma kusuru olan uzun QT sendromu tip 2 hastalarının tedavisinde değerlendirilmektedir. Serum glukokortikoid regüle kinaz 1, kardiyak sodyum kanallarını düzenler ve inhibisyonu özellikle uzun QT sendromu tip 2 ve tip 3 modellerinde ventriküler repolarizasyonu kısaltır80,81. Bu yeni tedavilerin klinik açıdan anlamlı kabul edilebilmesi için QTc değişikliğinin yalnızca doğru yönde olması değil, aynı zamanda klinik olarak anlamlı büyüklüğe ulaşması gerekir (örn. >40 ms kısalma)7,54.
Tedavi Yönetimi
Yerleşik tedavilerin doğrudan uygulanmasının ötesinde, uzun QT sendromunun yönetimi önemli ölçüde rafine edilmiştir. Genetik test, tanıyı doğrulama ve gen-özgül tedavi için ek yönlendirme sağlar40 (Tablo 2); ancak uzun QT sendromu olan hastaların %10–15’inde genetik tarama sonuçları negatiftir; bu durum, bu hastalarda aritmi riski ve tedavi yaklaşımları konusunda soru işaretlerine yol açmaktadır. 832 hastadan elde edilen veriler, genotip-negatif–fenotip-pozitif hastaların, genotip-pozitif–fenotip-pozitif hastalarla aynı şekilde tedavi edilmesi gerektiğini, çünkü aritmik risklerinin benzer olduğunu göstermiştir25. Genetik tarama sonuçları negatif veya belirsiz olduğunda, uzun QT sendromu tanısının doğru olduğundan emin olmak için her türlü çaba gösterilmelidir.
Tanı teyidi şu yollarla desteklenmelidir:
- Efor testi sırasında QTc davranışının değerlendirilmesi,
- 12 derivasyonlu Holter kaydı,
- T dalgasında çentikli, bifazik yapı veya T-dalgası alternansı gelişiminin incelenmesi,
- Ebeveynlerin tam kardiyak değerlendirmesi,
- Fiziksel olarak aktif hastalarda(yoğun egzersiz yapan), egzersize bağlı QTc uzamasını dışlamak için bir süre antrenman bırakma (detraining) önerilmesi83.
Önemli klinik yönetim unsurlarının çoğu genotipten bağımsızdır. Uzun QT sendromu dinamik bir durumdur; aritmi riski zaman içinde değişebilir ve bu nedenle tıbbi tedavinin optimize edilmesini gerektirebilir. Bu genellikle beta-blokerlere mexiletin veya sol kardiyak sempatik denervasyonun eklenmesi (yani üçlü tedavi) veya ICD implantasyonu anlamına gelir. Hastaların, tedavi optimizasyonuna olanak tanımak için en az yılda bir kez izlem ziyareti alması gerekir. Uzun QT sendromlu optimal medikal tedavi alan 946 hastanın uzun dönem sonuçlarını değerlendiren çalışmada, risk sınıflama skorlarına göre6,67 946 hastanın 142’sine ICD implante edilmesi gerektiği görüldü ancak ICD yalnızca 22 hastaya takılmıştı. ICD takılan hastaların yalnızca 3’ü uygun şok aldı ve hiçbir hasta ölmedi veya kardiyak arrest geçirmedi. Buna karşılık, takip sırasında bazı hastalarda artan aritmi riski belirdi ve bu hastalarda tedavi yoğunlaştırılarak aritmik olayların gelişmesi önlendi. Risk skorlarının, hastanın ilk başvurusunda veya tedavi başlanmadan önce uygulanması potansiyel olarak yanlıştır84 çünkü bu yaklaşım aşırı ve gerekçesiz ICD kullanımına yol açabilir56.Tedavinin başlatılması, aritmi gelişme eğilimini değiştirdiği için, tedavi optimizasyonundan sonra risk yeniden değerlendirilmeden ICD implantasyonu kararı vermek haklı gösterilemez85.Nitekim, uzun QT sendromlu 2861 hastadan elde edilen güncel veriler, kılavuzlara54 göre ICD implantasyonu adayı olanların yalnızca küçük bir kısmının gerçekten ICD’ye ihtiyaç duyduğunu göstermiştir78. Uzun QT sendromu olan genç hastalar için özellikle önemli bir konu, spor faaliyetlerine katılımdır; bu durum psikolojik açıdan büyük önem taşır. Başlangıçta oldukça koruyucu yaklaşım, özellikle uzun QT sendromu tip 2 ve tip 3 hastalarında giderek daha esnek bir yaklaşıma doğru evrilmektedir86. Bununla birlikte, bazı Avrupa ülkelerinde spor katılımının yasal düzenlemelerle kontrol edildiği ve spor hekimlerinin bunları göz ardı edemeyeceği unutulmamalıdır.
Ayrıca şu noktalara dikkat edilmelidir:
- En zayıf etkili beta-blokerlerin veya plasebo dozlarının reçete edilmesinden kaçınılmalıdır,
- Çok az sayıda hastada bilateral denervasyon gerekebilir; ancak sol kardiyak sempatik denervasyonun yeterli olup olmadığı değerlendirilmeden uygulanması uygun değildir87
- Epikardiyal kateter ablasyonu tedavi olarak önerilmiş olsa da88, ikna edici kanıt yetersizliği nedeniyle güçlü şekilde önerilmemektedir89.
Gerçekten de günümüzde mevcut olan ve uzun dönem boyunca son derece etkili ve güvenli olan tedaviler, zayıf gerekçeli deneysel yaklaşımlar için çok az alan bırakmaktadır.
Gen Tedavisi
Gen tedavisinin, uzun QT sendromu olan hastaların tedavisinde yararlı olabilme olasılığı yüksektir ve büyük bir ilgi alanıdır. Ancak, mevcut yaklaşımların tümü uygulanabilir değildir90. Uzun QT sendromu çoğunlukla, iyon kanal fonksiyonunu farklı şekillerde etkileyen tek nükleotid varyantlarını içerir ve başarılı bir gen tedavisi yaklaşımı ya varyant aleli susturmalı ya da doğrudan düzenleme yoluyla spesifik varyantı düzeltmelidir90. Gen susturma yaklaşımı; başlıca küçük RNA’lar olmak üzere çeşitli nükleik asitleri kullanarak, patojenik varyantın bulunduğu bölgeyi hedef alır ve varyant alelin ekspresyonunu bloke eder. Bu yaklaşım, uzun QT sendromu tip 1 ve tip 2 hücresel modellerinde in vitro olarak başarıyla uygulanmıştır;91-93 ancak en büyük kısıtı varyant-özgül olmasıdır ve uzun QT sendromuna yol açan yüzlerce varyantın varlığı, klinik uygulamadaki yaygın kullanımını önemli ölçüde sınırlar. Aynı sınırlılık doğrudan düzenleme (gene editing) yaklaşımları için de geçerlidir. Yakın dönemde geliştirilen baskılama–yerine koyma (suppression–replacement) terapisi90,94 uzun QT sendromu fenotipini, hastalığa neden olan varyanttan bağımsız olarak ,uzun QT sendromu tip 1’in94 ve tip 2’nin90 farklı hücresel modellerinde başarıyla düzeltmiş, böylece diğer yaklaşımların önemli bir sınırlılığını aşmıştır; ayrıca uzun QT sendromu tavşan modelinde de doğrulanmıştır95.Daha yakın zamanda aynı strateji kalmodulinopatilerin tedavisinde de uygulanmıştır96. Bununla birlikte, uzun QT sendromunda gen tedavisinin klinik pratiğe başarılı biçimde aktarılması ve uygulanabilmesi için halen aşılması gereken önemli zorluklar vardır. Baskılama yerine koyma terapisinde, verilmesi gereken doğru dozun belirlenmesini gerektirir; çünkü yetersiz tedavi uzun QT fenotipini düzeltmezken, aşırı tedavi proaritmik olabilir. Gen tedavileri, genellikle homojen olmayan düzeyde transdükte hücre popülasyonları üretir ve repolarizasyon heterojenitesinin artması nedeniyle potansiyel proaritmi riski taşır. Gen tedavisi uygulanan hastalarda bildirilmiş bazıları ölümcül advers olaylar nedeniyle güvenliğe ilişkin endişeler mevcuttur.97-99. Ayrıca, gen tedavisinin uygulanmasına ilişkin bir diğer önemli husus, uzun QT sendromunda aritmi riskinin zamanla artmaması ve mevcut tedavilerin son derece düşük mortalite ile ilişkili olmasıdır56,78. Güncel tedavilerin etkinliği, gen tedavisi gibi deneysel yaklaşımlara ihtiyaç duyan hasta sayısını büyük ölçüde azaltmaktadır; bu tür yaklaşımlar, çok yüksek riskli hastalar için daha uygun olabilir (örn. yaşamın ilk yılında kardiyak olay geçiren bebekler; bazı kalmodulin varyantları19 veya ağır seyirli hastalığa yol açan varyantlar [örn. SCN5A’daki p.R1623Q varyantı] bulunan ve tam tedaviye rağmen uygun ICD şokları almaya devam eden hastalar).41,56
Edinilmiş Uzun QT Sendromu
QT aralığı; hipokalemi, bradikardi, kalp bloğu100 ve özellikle IKr bloklayıcı etkiye sahip ilaçların kullanımı gibi çeşitli durumlarda uzayabilir101. Edinilmiş uzun QT sendromu klinik açıdan önemlidir, çünkü torsades de pointes ve ani kardiyak ölüm için anlamlı bir risk taşır102. Soruna yol açan etkenin ortadan kaldırılması, tekrarların önlenmesini sağlar.
Edinilmiş uzun QT sendromunun gelişme olasılığı, iki ana faktöre bağlıdır:
Kullanılan ilacın doğasında bulunan intrinsik risk — esas olarak IKr blokajının derecesine bağlıdır,
Bireyin repolarizasyon rezervi düzeyi,103 ki bu da genetik faktörler tarafından modüle edilir 104-106
Bu genetik yatkınlık; çok nadir yaygın genetik varyantları içerir104-106.
Edinilmiş uzun QT sendromu olan hastalarda patojenik veya olası patojenik varyant saptanma olasılığı başlıca üç değişkene bağlıdır:
- Yaşın 40’ın altında olması,
- Başlangıç QTc değerinin 440 ms’nin üzerinde olması,
- Aritmik epizotların varlığı104
Bu değişkenler, bazı durumlarda edinilmiş uzun QT sendromunun, düşük penetranslı latent konjenital uzun QT sendromunu ortaya çıkarabileceğini düşündürmektedir bu durum ilk kez 1982’de öne sürülmüştür107.
Bu faktörlerin varlığında, edinilmiş uzun QT sendromu olan hastalara hastalıkla ilişkili genler için moleküler genetik test önerilmelidir23. Buna ek olarak, fonksiyonel etkisi olan iki nadir varyant KCNE1’de p.D85N ve SCN5A’da p.S1103Y edinilmiş uzun QT sendromu ile tutarlı biçimde ilişkilidir108,109. QT aralığını etkileyen 61 yaygın genetik varyantın kombinasyonu, edinilmiş uzun QT sendromundaki değişkenliğin %30’una kadarını açıklayabilmektedir106. Yaygın varyantların rutin klinik amaçlı test edilmesi, araştırma ortamı dışına halen önerilmemektedir. Edinilmiş uzun QT sendromu, genetik ve edinilmiş faktörlerin birlikte repolarizasyon rezervini bozarak aritmik olayları tetikleyebileceğinin tipik bir örneğidir. Aşırı fiziksel antrenman, özellikle ergenlerde, uzun QT sendromunu taklit eden belirgin QT uzamasına yol açabilir ve bu durum uzun süreli sonuçları olan tanısal hatalara zemin hazırlayabilir83. Bu kişiler genellikle:
Uzun QT sendromu için aile öyküsüne sahip değildir,
- Asemptomatiktir,
- Genotip-negatiftir.
Bu ventriküler repolarizasyon anormallikleri, 3–4 aylık egzersiz bırakma (detraining) ile geri dönüşlüdür83. Bu anormalliklerin aritmojenik potansiyeli tam olarak bilinmemekle birlikte, QT uzamasını önlemek için antrenman şiddetinin azaltılması gerekir. Spor hekimlerinin, genç bireyleri erken yaşta uzun QT sendromu tanısıyla etiketlemekten kaçınmak için bu fenomenin farkında olması önemlidir.
Sonuçlar
Uzun QT sendromu, sıklıkla ölümcül seyredebilmesine rağmen günümüzde etkili ve güvenli tedavilerin bulunduğu bir hastalıktır ve bu sayede hastaların neredeyse tamamında normal bir yaşam kalitesi sağlanabilmektedir. Sendromun doğru şekilde yönetilebilmesi özel bir uzmanlık gerektirir ve klinisyenler, hastalığın varlığından şüphelenebilmeli ve hastaları, uzun QT sendromu konusunda özel deneyime sahip yüksek hacimli merkezlere yönlendirebilmelidir.
Kaynakça;
References
1. Grant RP. Clinical electrocardiography: the spatial vector approach. New York: McGraw-Hill, 1957.
2. Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957; 54: 59-68.
3. Romano C, Gemme G, Pongiglione R. Rare cardiac arrhythmias of the pediatric age. I. Repetitive paroxysmal tachycardia. Minerva Pediatr 1963; 15: 1155-64. (In Italian.)
4. Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc 1964; 54: 103-6.
5. Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J 1975; 89: 378-90.
6. Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence of the congenital long- QT syndrome. Circulation 2009; 120: 1761-7.
7. Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J 2013; 34: 3109-16.
8. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995; 80: 795-803.
9. Wang Q, Shen J, Li Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 1995; 4: 1603-7.
10. Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996; 12: 17-23.
11. Schwartz PJ, Ackerman MJ, Antzelevitch C, et al. Inherited cardiac arrhythmias. Nat Rev Dis Primers 2020; 6: 58.
12. Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 1997; 15: 186-9.
13. Schulze-Bahr E, Wang Q, Wedekind H, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet 1997; 17: 267-8.
14. Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 2006; 113: 783-90.
15. Adler A, Novelli V, Amin AS, et al. An international, multicentered, evidencebased reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020; 141: 418-28.
16. Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119: 19-3 17. Schwartz PJ, Crotti L, Nyegaard M, Overgaard MT. Role of calmodulin in cardiac disease: insights on genotype and phenotype. Circ Genom Precis Med 2024; 17(5): e004542.
18. Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013; 127: 1009-17.
19. Crotti L, Spazzolini C, Nyegaard M, et al. Clinical presentation of calmodulin mutations: the International Calmodulinopathy Registry. Eur Heart J 2023; 44: 3357- 70.
20. Bains S, Dotzler SM, Krijger C, et al. A phenotype-enhanced variant classification framework to decrease the burden of missense variants of uncertain significance in type 1 long QT syndrome. Heart Rhythm 2022; 19: 435-42.
21. Neves R, Crotti L, Bains S, et al. Phenotype- enhanced variant classification framework to decrease the burden of VUS in LQTS type 2. JACC Clin Electrophysiol (in press).
22. O’Neill MJ, Ng C-A, Aizawa T, et al. Multiplexed assays of variant effect and automated patch clamping improve KCNH2-LQTS variant classification and cardiac event risk stratification. Circulation 2024; 150: 1869-81.
23. Wilde AAM, Semsarian C, Márquez MF, et al.. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/ Latin American Heart Rhythm Society
(LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace 2022; 24: 1307-67.
24. Lahrouchi N, Tadros R, Crotti L, et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT Syndrome. Circulation 2020; 142: 324-38.
25. Shimamoto K, Dagradi F, Ohno S, et al. Clinical features, long-term prognosis, and clinical management of genotypenegative long QT syndrome patients. JACC Clin Electrophysiol 2024; 10: 2584- 96.
26. Brink PA, Crotti L, Corfield V, et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 2005; 112: 2602-10.
27. Schwartz PJ, Crotti L, George AL Jr. Modifier genes for sudden cardiac death. Eur Heart J 2018; 39: 3925-31.
28. Crotti L, Monti MC, Insolia R, et al. NOS1AP is a genetic modifier of the long- QT syndrome. Circulation 2009; 120: 1657- 63.
29. Lee Y-K, Sala L, Mura M, et al. MTMR4 SNVs modulate ion channel degradation and clinical severity in congenital long QT syndrome: insights in the mechanism of action of protective modifier genes. Cardiovasc Res 2021; 117: 767-79. 30. Ronchi C, Bernardi J, Mura M, et al. NOS1AP polymorphisms reduce NOS1 activity and interact with prolonged repolarization in arrhythmogenesis. Cardiovasc Res 2021; 117: 472-83.
31. Bazett HC. An analysis of the timerelations of electrocardiograms. Heart 1920; 7: 353-70.
32. Stramba-Badiale M, Karnad DR, Goulene KM, et al. For neonatal ECG screening there is no reason to relinquish old Bazett’s correction. Eur Heart J 2018; 39: 2888-95.
33. Schwartz PJ, Garson A Jr, Paul T, et al. Guidelines for the interpretation of the neonatal electrocardiogram. Eur Heart J 2002; 23: 1329-44.
34. Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med 2003; 348: 1866- 74.
35. Nador F, Beria G, De Ferrari GM, et al. Unsuspected echocardiographic abnormality in the long QT syndrome: diagnostic, prognostic, and pathogenetic implications. Circulation 1991; 84: 1530-42.
36. Haugaa KH, Edvardsen T, Leren TP, Gran JM, Smiseth OA, Amlie JP. Left ventricular mechanical dispersion by tissue Doppler imaging: a novel approach for identifying high-risk individuals with long QT syndrome. Eur Heart J 2009; 30: 330-7.
37. ter Bekke RMA, Haugaa KH, van den Wijngaard A, et al. Electromechanical window negativity in genotyped long-QT syndrome patients: relation to arrhythmia risk. Eur Heart J 2015; 36: 179-86.
38. Malfatto G, Beria G, Sala S, Bonazzi O, Schwartz PJ. Quantitative analysis of T wave abnormalities and their prognostic implications in the idiopathic long QT syndrome. J Am Coll Cardiol 1994; 23:
296-301.
39. Schwartz PJ, Malliani A. Electrical alternation of the T-wave: clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long Q-T syndrome. Am Heart J 1975; 89: 45-50.
40. Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation
2001; 103: 89-95.
41. Spazzolini C, Mullally J, Moss AJ, et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol 2009; 54: 832-7.
42. Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med 2000; 343: 262-7.
43. Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation 2007; 115: 361-7.
44. Crotti L, Tester DJ, White WM, et al.Long QT syndrome-associated mutations in intrauterine fetal death. JAMA 2013; 309: 1473-82.
45. Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 1998; 338: 1709-14.
46. Saul JP, Schwartz PJ, Ackerman MJ, Triedman JK. Rationale and objectives for ECG screening in infancy. Heart Rhythm 2014; 11: 2316-21.
47. Schwartz PJ, Crotti L, Nyegaard M, Overgaard MT. Calmodulin, sudden death, and the Folbigg case: genes in court. Eur Heart J 2024; 45: 1801-3.
48. Boeri C, Sarto P, Cerea P, et al. TPfusion at peak exercise: a novel marker for the recognition of unsuspected long QT syndrome patients. Europace 2025; 27.
49. Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 2011; 124: 2181-4.
50. Abrahams T, Davies B, Laksman Z, et al. Provocation testing in congenital long QT syndrome: a practical guide. Heart Rhythm 2023; 20: 1570-82.
51. Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation 2011; 124: 2187-94.
52. Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol 2010; 55: 1955-61.
53. Ackerman MJ, Khositseth A, Tester DJ, Hejlik JB, Shen W-K, Porter C. Epinephrine- induced QT interval prolongation: a gene-specific paradoxical response in congenital long QT syndrome. Mayo Clin Proc 2002; 77: 413-21.
54. Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2022; 43: 3997- 4126.
55. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000; 101: 616-23.
56. Dusi V, Dagradi F, Spazzolini C, et al. Long QT syndrome: importance of reassessing arrhythmic risk after treatment initiation. Eur Heart J 2024; 45: 2647-56.
57. Wilde AAM, Moss AJ, Kaufman ES, et al. Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation 2016; 134: 872-82.
58. Chockalingam P, Crotti L, Girardengo G, et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J Am Coll Cardiol 2012; 60: 2092-9.
59. Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in longQT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures.” Circulation 2009; 119: 215-21.
60. Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm 2009; 6: 752-9.
61. Schwartz PJ. Cardiac sympathetic denervation to prevent life-threatening arrhythmias. Nat Rev Cardiol 2014; 11: 346-53.
62. Schwartz PJ, Ackerman MJ. Cardiac sympathetic denervation in the prevention of genetically mediated life-threatening ventricular arrhythmias. Eur Heart J 2022; 43: 2096-102.
63. Dusi V, Pugliese L, De Ferrari GM, et al. Left cardiac sympathetic denervation for long QT syndrome: 50 years’ experience provides guidance for management. JACC Clin Electrophysiol 2022; 8: 281-94.
64. Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am J Cardiol 1976; 37: 1034-40.
65. Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation 2004; 109: 1826-33.
66. Niaz T, Bos JM, Sorensen KB, Moir C, Ackerman MJ. Left cardiac sympathetic denervation monotherapy in patients with congenital long QT syndrome. Circ Arrhythm Electrophysiol 2020; 13(12): e008830.
67. Schwartz PJ, Spazzolini C, Priori SG, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter- defibrillator and what happens to them? Data from the European Long-QT
Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD) Registry. Circulation 2010; 122: 1272-82.
68. Antiel RM, Bos JM, Joyce DD, et al. Quality of life after videoscopic left cardiac sympathetic denervation in patients with potentially life-threatening cardiac channelopathies/cardiomyopathies. Heart Rhythm 2016; 13: 62-9.
69. Olde Nordkamp LRA, Driessen AHG, Odero A, et al. Left cardiac sympathetic denervation in the Netherlands for the treatment of inherited arrhythmia syndromes. Neth Heart J 2014; 22: 160-6.
70. Li K, Yang J, Guo W, et al. Videoassisted thoracoscopic left cardiac sympathetic denervation in Chinese patients with long QT syndrome. Int Heart J 2018; 59: 1346-51.
71. Schwartz PJ. Efficacy of left cardiac sympathetic denervation has an unforeseen side effect: medicolegal complications. Heart Rhythm 2010; 7: 1330-2.
72. Schwartz PJ, Ackerman MJ. Implantable cardioverter defibrillators for long QT syndrome and catecholaminergic polymorphic ventricular tachycardia? (Not so fast, Louis). Europace 2025 October 18 (Epub
ahead of print).
73. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate:
implications for gene-specific therapy. Circulation 1995; 92: 3381-6.
74. Mazzanti A, Maragna R, Faragli A, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol 2016; 67: 1053-8.
75. Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 2007; 116: 1137-44.
76. Crotti L, Neves R, Dagradi F, et al. Therapeutic efficacy of mexiletine for long QT syndrome type 2: evidence from human induced pluripotent stem cell-derived cardiomyocytes, transgenic rabbits, and
patients. Circulation 2024; 150: 531-43.
77. Schwartz PJ, Spazzolini C. Are long QT syndrome patients managed differently in different countries? Eur Heart J 2025; 46: 3467-9.
78. Neves R, Crotti L, Bains S, et al. Frequency of and outcomes associated with nonadherence to guideline-based recommendations for an implantable cardioverter defibrillator in patients with congenital
long QT syndrome. Heart Rhythm 2025; 22: 2073-81.
79. Mehta A, Ramachandra CJA, Singh P, et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur Heart J 2018; 39: 1446-55.
80. Kim M, Sager PT, Tester DJ, et al. SGK1 inhibition attenuates the action potential duration in reengineered heart cell models of drug-induced QT prolongation. Heart Rhythm 2023; 20: 589-95.
81. Giannetti F, Barbieri M, Shiti A, et al. Gene- and variant-specific efficacy of serum/ glucocorticoid-regulated kinase 1 inhibition in long QT syndrome types 1 and 2. Europace 2023; 25: euad094.
82. Schwartz PJ, Gnecchi M, Dagradi F, et al. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome type 2. Eur Heart J 2019; 40: 1832-6.
83. Dagradi F, Spazzolini C, Castelletti S, et al. Exercise training-induced repolarization abnormalities masquerading as congenital long QT syndrome. Circulation 2020; 142: 2405-15.
84. Corrado D, Link MS, Schwartz PJ. Implantable defibrillators in primary prevention of genetic arrhythmias: a shocking choice? Eur Heart J 2022; 43: 3029-40.
85. Wilde AAM, van der Werf C. Risk scores in congenital long QT syndrome: friend or foe? Eur Heart J 2024; 45: 2657-9.86. Lampert R, Day S, Ainsworth B, et al. Vigorous exercise in patients with congenital
long QT syndrome: results of the prospective, observational, multinational LIVE-LQTS study. Circulation 2024; 150: 516-30.
87. Akkuş M, Seyrek Y, Kafalı HC, Ergül Y. Bilateral cardiac sympathetic denervation in children with long-QT syndrome and catecholaminergic polymorphic ventricular tachycardia. J Electrocardiol 2020; 61: 32-6.
88. Pappone C, Ciconte G, Anastasia L, et al. Right ventricular epicardial arrhythmogenic substrate in long-QT syndrome patients at risk of sudden death. Europace 2023; 25: 948-55.
89. Wilde AAM, Ackerman MJ. Counterpoint: ablation in long QT syndrome. Heart Rhythm 2023; 20: 1785-6.
90. Bains S, Giudicessi JR, Odening KE, Ackerman MJ. State of gene therapy for monogenic cardiovascular diseases. Mayo Clin Proc 2024; 99: 610-29.
91. Matsa E, Dixon JE, Medway C, et al. Allele-specific RNA interference rescues the long-QT syndrome phenotype in humaninduced pluripotency stem cell cardiomyocytes. Eur Heart J 2014; 35: 1078-87.
92. Limpitikul WB, Dick IE, Tester DJ, et al. A precision medicine approach to the rescue of function on malignant calmodulinopathic Long-QT Syndrome. Circ Res 2017; 120: 39-48.
93. Ge N, Liu M, Li R, et al. Using ribonucleoprotein- based CRISPR/Cas9 to edit single nucleotide on human induced pluripotent stem cells to model type 3 Long QT Syndrome (SCN5A). Stem Cell Rev Rep
2023; 19: 2774-89.
94. Dotzler SM, Kim CSJ, Gendron WAC, et al. Suppression-replacement KCNQ1 gene therapy for type 1 Long QT Syndrome. Circulation 2021; 143: 1411-25.
95. Bains S, Giammarino L, Nimani S, et al. KCNQ1 suppression-replacement gene therapy in transgenic rabbits with type 1 long QT syndrome. Eur Heart J 2024; 45: 3751-63.
96. Hamrick SK, Kim CSJ, Tester DJ, et al. Single construct suppression and replacement gene therapy for the treatment of all CALM1-, CALM2-, and CALM3-mediated arrhythmia disorders. Circ Arrhythm Electrophysiol 2024; 17(8): e012036.
97. Somia N, Verma IM. Gene therapy: trials and tribulations. Nat Rev Genet 2000; 1: 91-9.
98. Wilson JM, Flotte TR. Moving forward after two deaths in a gene therapy trial of myotubular myopathy. Hum Gene Ther 2020; 31: 695-6.
99. Rocket pharmaceuticals provides update on phase 2 clinical trial of RP-A501 for Danon disease. May 27, 2025 (https:// ir .rocketpharma.com/news-releases/news release-details/rocket-pharmaceuticals-provides
update-phase-2-clinical-trial-rp).
100. Vos MA, de Groot SH, Verduyn SC, et al. Enhanced susceptibility for acquired torsade de pointes arrhythmias in the dog with chronic, complete AV block is related to cardiac hypertrophy and electrical remodeling. Circulation 1998; 98: 1125-35.
101. Schwartz PJ, Woosley RL. Predicting the unpredictable: drug-induced QT prolongation and torsades de pointes. J Am Coll Cardiol 2016; 67: 1639-50. 102. Roden DM. Long-QT syndrome. N Engl J Med 2008; 358: 169-76.
103. Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol 1998; 21: 1029-34.
104. Itoh H, Crotti L, Aiba T, et al. The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur Heart J 2016; 37: 1456-64.
105. Giudicessi JR, Roden DM, Wilde AAM, Ackerman MJ. Classification and reporting of potentially proarrhythmic common genetic variation in long QT syndrome genetic testing. Circulation 2018; 137: 619-30.
106. Strauss DG, Vicente J, Johannesen L, et al. Common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk: a pilot study. Circulation 2017; 135: 1300-10.
107. Moss AJ, Schwartz PJ. Delayed repolarization (QT or QTU prolongation) and malignant ventricular arrhythmias. Mod Concepts Cardiovasc Dis 1982; 51: 85-90.
108. Kääb S, Crawford DC, Sinner MF, et al. A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet 2012; 5: 91-9.
109. Akylbekova EL, Payne JP, Newton- Cheh C, et al. Gene-environment interaction between SCN5A-1103Y and hypokalemia influences QT interval prolongation in African Americans: the Jackson Heart Study. Am Heart J 2014; 167(1): 116-122.e1.